Characteristics/Phenotype
Wolf-Hirschhorn syndrome in 1961. They
described a child with midline fusion defects, and subsequent cytogenetic
studies revealed a chromosomal deletion of the short arm of chromosome 4. Wolf-Hirschhorn syndrome (WHS) refers to a condition that is
caused by a missing part (deletion) of the short arm of chromosome 4. This
missing genetic material results in severe developmental delays, a
characteristic facial appearance, and may include a variety of other birth
defects. The short arm of a chromosome is called the “p” arm. Thus, this
syndrome is also known as 4p-syndrome or deletion 4p syndrome, and occasionally
as Wolf syndrome.
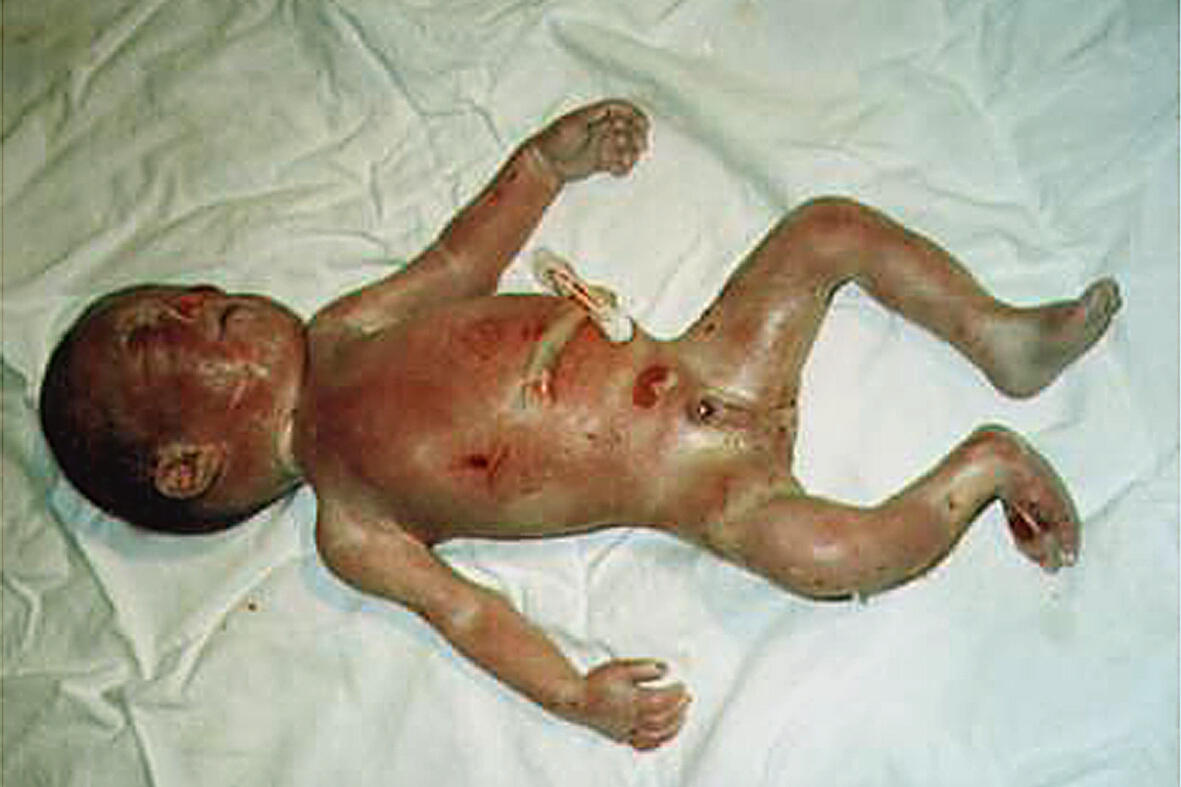
Almost everyone with this
disorder has distinctive facial features, including a broad, flat nasal bridge
and a high forehead. The eyes are widely spaced and may be protruding. Other
characteristic facial features include a shortened distance between the nose
and upper lip (a short philtrum), a downturned mouth, a small chin
(micrognathia), and poorly formed ears with small holes (pits) or flaps of skin
(tags). Additionally, affected individuals may have asymmetrical facial
features and an unusually small head (microcephaly).
People with
Wolf-Hirschhorn syndrome experience delayed growth and development. Slow growth
begins before birth, and affected infants tend to have problems feeding and
gaining weight (failure to thrive). They also have weak muscle tone (hypotonia)
and underdeveloped muscles. Motor skills such as sitting, standing, and walking
are significantly delayed. Most children and adults with this disorder also
have short stature.
Intellectual disability
ranges from mild to severe in people with Wolf-Hirschhorn syndrome. Compared to
people with other forms of intellectual disability, their socialization skills
are strong, while verbal communication and language skills tend to be weaker.
Most affected children also have seizures, which may be resistant to treatment.
Seizures tend to disappear with age.
Additional features of
Wolf-Hirschhorn syndrome include skin changes such as mottled or dry skin,
skeletal abnormalities such as abnormal curvature of the spine (scoliosis and
kyphosis), dental problems including missing teeth, and an opening in the roof
of the mouth (cleft palate) and/or in the lip (cleft lip). Wolf-Hirschhorn
syndrome can also cause abnormalities of the eyes, heart, genitourinary tract,
and brain.
A condition called
Pitt-Rogers-Danks syndrome has features that overlap with those of
Wolf-Hirschhorn syndrome. Researchers now recognize that these two conditions
are actually part of a single syndrome with variable signs and symptoms.
It has been estimated that approximately 35% of
individuals who have WHS die within the first two years of life. Many individuals
who have WHS survive to adulthood. Universally, children with WHS have severe
or profound developmental delays, however, there are many affected individuals
who are able to walk and some that are able to talk in short sentences.
Frequency
The
prevalence of Wolf-Hirschhorn syndrome is estimated to be 1 in 50,000 births.
However, this may be an underestimate because it is likely that some affected
individuals are never diagnosed.
For
unknown reasons, Wolf-Hirschhorn syndrome occurs in about twice as many females
as males.
Diagnosis
When WHS is suspected, chromosome
analysis should be performed and the laboratory should be informed as to what
syndrome is suspected. If the deletion is not visible, then fluorescent in situ
hybridization (FISH) can be done specifically for the critical 4p16.3 region of
chromosome 4.
Interestingly, there is a syndrome
called Pitt-Rogers-Danks syndrome (PRDS) that has been reported to have similar
characteristics to WHS. Several individuals who have initially been diagnosed
with PRDS subsequently had FISH analysis that detected a deletion of 4p, and
thus the individuals were reclassified as having WHS. Some feel that PRDS is
actually WHS without obvious deletions of 4p.
When a couple has had a child diagnosed
to have WHS, and a member of that couple carries a balanced
translocation, genetic counseling should be offered to discuss
reproductive options.
If ultrasound examination reveals
findings consistent with the possibility of WHS in a family with no history of
WHS, genetic counseling and prenatal diagnosis should be offered.
Causes
Wolf-Hirschorn is caused by a
chromosomal deletion that occurs as a random (de novo) event during the
formation of reproductive cells (eggs or sperm) or in early embryonic
development. More complex chromosomal rearrangements can also occur as de novo
events, which may help explain the variability in the condition's signs and
symptoms. De novo chromosomal changes occur in people with no history of the
disorder in their family.
A small percentage of all people with
Wolf-Hirschhorn syndrome have the disorder as a result of an unusual
chromosomal abnormality such as a ring chromosome 4. Ring chromosomes occur
when a chromosome breaks in two places and the ends of the chromosome arms fuse
together to form a circular structure. In the process, genes near the ends of
the chromosome are lost.
In the remaining cases of
Wolf-Hirschhorn syndrome, an affected individual inherits a copy of chromosome
4 with a deleted segment. In these cases, one of the individual's parents
carries a chromosomal rearrangement between chromosome 4 and another
chromosome. This rearrangement is called a balanced translocation.
Between 85 and 90
percent of all cases of Wolf-Hirschhorn syndrome are not inherited.
Treatment
There is no treatment for
the underlying condition of WHS. Treatment and management for patients who have
WHS are specific to each individual. For example, some individuals who have WHS
may have heart defects or a cleft lip and/or palate that may require surgery,
while others may not. Therefore, there is no specific treatment for individuals
who have WHS, rather, the treatment and management is geared toward that
particular individual’s needs and is likely to include several medical
specialists. Physiotherapy and occupational therapy are recommended. Some
patients require physical aids, e.g. wheel chair, splints, hearing aids etc.
Patients with congenital heart defects, clubfeet, and cryptorchidism have to be
surgically treated. Those with seizures need
recurrent EEGs and antiepileptic drugs. Information about patients who
have WHS has been compiled and provides a comprehensive look into the natural
history of this condition and the needed management. The collection of this
information has shown that many of these individuals may achieve more development
than was previously believed possible.
The following management
recommendations have been made by Drs. Battaglia and Carey @
http://wolfhirschhorn.org/about-wolf-hirschhorn-syndrome/:
- Feeding problems should be addressed and may require interventionsuch
as placement of a gastrostomy tube.
- Characterization of seizures is important and
treatment with antiepileptic medications such as valproic acid should
be investigated and may help control the seizure activity in many
individuals.
- Skeletal abnormalities such as clubfoot
should be addressed and treatment should be considered. It should not be
assumed that clubfoot does not need addressed because the child will never
walk. Children with WHS have learned to walk unassisted.
- As approximately 30% of individuals may have
congenital heart defects, the heart should be examined.
- Hearing loss may occur and because some
children are able to learn to talk in short sentences, they should
be screened for hearing problems.
Eye abnormalities
may be present and thus an ophthalmology exam should be performed to rule
out any eye problems, even if no obvious signs are present.
Advices
In
regards to the development of patients with WHS, it is suggested that
individuals participate in personal development programs to assist with social
and occupational therapy for motor skills.
References
http://ghr.nlm.nih.gov/condition/wolf-hirschhorn-syndrome
Photos from:
https://blogger.googleusercontent.com/img/b/R29vZ2xl/AVvXsEjXrFTNKz9va7SlCZN7SwWQGlLC4dyQzml6SHy9fGhUNhOYuTvHVphDOP97vNPInVXAb-Lw3CUY8b_V5F3uaXkefi2B1hh-1ywWjuuYfRShIqHdoSsJasV2DBsabzze0ocyI-Q9Su8PTdyh/s1600/DSC_0033soft.jpg
http://upload.wikimedia.org/wikipedia/commons/f/ff/Wolf-hirschhorn.jpg



.jpg)


.jpg)



